
Congenital
Surgical correction of congenital eyelid and orbital conditions present from birth — ptosis, blocked tear ducts, hemangiomas, and more.
Congenital Eyelid & Orbital Conditions
Congenital conditions of the eyelid, orbit, and lacrimal system are present from birth or become apparent in early childhood. They range from minor cosmetic variations to conditions that threaten vision through amblyopia — the brain’s suppression of input from an eye whose image is chronically blurred, blocked, or misaligned. Early detection and appropriately timed surgery are critical for visual development.
Dr. Brown cares for children with eyelid and orbital conditions that require surgical expertise, coordinating with pediatric ophthalmology for vision assessment and amblyopia management. The sections below describe the most common congenital conditions seen in this practice.
Congenital Ptosis
Congenital ptosis is a drooping of the upper eyelid present from birth, most commonly due to a developmental dysgenesis of the levator palpebrae superioris muscle. Unlike acquired ptosis in adults, in which the muscle is normal but its tendon is stretched, congenital ptosis is caused by fibrofatty replacement of levator muscle fibers — producing a weak, non-elastic muscle that neither lifts well nor relaxes fully.
Why Timing Matters
The visual system develops in the first 7–10 years of life. During this critical period, both eyes must deliver a clear, focused image to the brain for normal visual development. Ptosis threatens vision in two ways:
- Deprivation amblyopia — a severely drooped lid that covers the pupil blocks all light from reaching the retina. This is the most urgent indication for early surgery, often within weeks of diagnosis
- Astigmatic amblyopia — even when the lid does not cover the pupil, its weight on the cornea induces astigmatism that blurs the image and can suppress vision in that eye
Children also compensate for ptosis by adopting a chin-up head posture or by raising the brow. These compensatory postures do not protect vision but indicate that the child is attempting to see under a drooped lid — a sign that the ptosis is visually significant.
Associated Conditions
- Superior rectus weakness — occurs in approximately 25% of congenital ptosis cases; influences surgical planning because frontalis sling must account for reduced upgaze
- Marcus Gunn jaw-wink — present in 2–13% of congenital ptosis; see the Ptosis page for detailed discussion
- Blepharophimosis syndrome — autosomal dominant condition with bilateral ptosis, horizontal lid shortening (phimosis), epicanthus inversus, and telecanthus; requires staged reconstruction
- Strabismus — present in approximately 30% of cases
Surgical Treatment
The surgical approach depends on the degree of levator function (how many millimeters the lid moves from down-gaze to up-gaze):
- Good levator function (≥ 5 mm) — levator resection or aponeurotic advancement shortens and tightens the levator; suitable for mild to moderate ptosis
- Poor levator function (< 4 mm) — frontalis sling (suspension) connects the lid to the frontalis muscle using a silicone rod, autologous fascia lata, or synthetic material. The child lifts the lid by raising the brow
Full details of ptosis evaluation and surgical technique are on the Ptosis page.
Congenital Nasolacrimal Duct Obstruction (CNLDO)
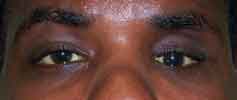
Congenital nasolacrimal duct obstruction is the most common cause of tearing and eye discharge in infants. Approximately 6% of newborns have an imperforate membrane at the distal nasolacrimal duct (the valve of Hasner) that normally opens at or shortly after birth. Tears that cannot drain stagnate and frequently become infected.
Presentation
- Persistent tearing (epiphora) beginning in the first weeks of life — typically unilateral
- Mucoid or mucopurulent discharge at the medial canthus
- Regurgitation of discharge when pressure is applied over the lacrimal sac (Crigler massage sign)
- Secondary skin maceration and redness at the medial canthus
- Episodes of conjunctivitis-appearing infection that temporarily improve with topical antibiotics but recur
Classification
CNLDO presents in one of four ways:
- Simple obstruction — the most common form; imperforate membrane at the valve of Hasner
- Congenital lacrimal fistula — an abnormal epithelial-lined opening from the lacrimal sac to the skin surface, visible as a small opening inferomedial to the medial canthus
- Congenital dacryocele (mucocele) — a cystic swelling of the lacrimal sac present at birth from obstructed drainage both proximally (via the canaliculi) and distally. The bluish-gray mass is medial and inferior to the medial canthus and is often confused with a hemangioma. Intranasal cysts may obstruct the airway
- Acute dacryocystitis — bacterial infection of an obstructed lacrimal sac; presents with erythema, warmth, and fluctuant swelling over the lacrimal sac; requires systemic antibiotics and urgent evaluation
Natural History and Non-Surgical Treatment
The vast majority of simple CNLDO resolves spontaneously: 90% open by 12 months of age. The accepted initial management is Crigler massage — digital compression over the lacrimal sac 5–10 times, 2–3 times daily, to create a hydrostatic pressure wave that opens the distal membrane. Topical antibiotics are used episodically for infectious exacerbations but do not cure the obstruction.
Surgical Treatment
If spontaneous resolution has not occurred by 9–12 months, probing is offered:
- Office probing (awake) — a fine metallic probe is passed through the punctum and canaliculus into the lacrimal sac and through the nasolacrimal duct under magnification; success rate ~75–90% for primary probing before age 12 months
- Probing under general anesthesia — performed for older children or when cooperation for awake probing is not feasible; allows irrigation and nasal endoscopy
- Silicone intubation — a silicone tube is threaded through the entire lacrimal drainage system and left in place 3–6 months; used for failed probing or complex anatomy
- Balloon dacryoplasty — a specialized balloon catheter is inflated within the duct to dilate it; used after failed probing
- Dacryocystorhinostomy (DCR) — creation of a new drainage channel from the lacrimal sac to the nasal cavity, bypassing the obstructed duct; reserved for cases failing all other measures. Full details on the Lacrimal System page
Congenital Orbital Conditions
The orbit develops from a complex interaction of neural crest cells, mesoderm, and optic vesicle signals. Disruption of these processes can result in abnormalities of orbital size, shape, and contents.
Microphthalmos and Anophthalmos
Microphthalmos (abnormally small eye) and anophthalmos (absent eye) represent a spectrum of severe developmental failures of the optic vesicle. They may be isolated or part of a systemic syndrome. Anophthalmos requires early intervention with conformers and orbital expanders to stimulate orbital growth — the orbital bones depend on the eye’s presence to grow normally. Without treatment, the orbit remains small and socket reconstruction in adulthood is challenging.
For detailed information about socket and anophthalmic orbital management, see the Anophthalmos page.
Dermoid Cysts
Dermoid cysts are the most common benign orbital mass of childhood — choristomas that arise at embryonic suture lines from trapped ectoderm. The superolateral orbital rim (frontozygomatic suture) is the classic location. They are smooth, non-tender, freely mobile masses that do not transilluminate. Most present in the first decade. Management is complete surgical excision — the cyst wall must be intact; rupture causes severe granulomatous inflammation. Deep dermoids extending intracranially require CT planning before surgery.
Capillary Hemangioma
The most common orbital tumor of infancy. Periorbital infantile hemangiomas may extend into the orbit, producing ptosis and proptosis. They proliferate rapidly in the first year of life, then involute slowly over years. Treatment with oral propranolol (1–3 mg/kg/day) is now first-line for lesions that threaten vision or cause significant deformity. See the Orbital Tumors page for full details.
Congenital Eyelid Anomalies
Blepharophimosis Syndrome
Blepharophimosis, ptosis, and epicanthus inversus syndrome (BPES) is an autosomal dominant condition caused by mutations in the FOXL2 gene. The defining features are:
- Blepharophimosis — horizontally shortened palpebral fissures (<25 mm, normal ≈28–30 mm)
- Ptosis — bilateral, with poor levator function; requires frontalis sling
- Epicanthus inversus — skin fold arising from the lower lid and partially covering the medial canthus
- Telecanthus — widened intercanthal distance from medial canthal tendon laxity
Surgical correction is staged: medial canthal tendon plication (Y-V or W-plasty) is performed at 12–18 months to widen the fissure and correct telecanthus before ptosis repair. Ptosis repair with frontalis sling typically follows. BPES Type I is associated with premature ovarian failure; genetic counseling and gynecologic follow-up are indicated for affected females.
Congenital Entropion and Epiblepharon
Epiblepharon is a horizontal fold of skin just below the lower lid margin that redirects lashes toward the cornea. It is particularly common in East Asian children. Most cases resolve spontaneously by age 5 as the face elongates and orbicularis atrophies. Surgery (excision of a skin-muscle strip) is indicated for persistent corneal staining, pain, or visual disturbance.
Congenital entropion is an inward rotation of the lid margin itself. It is less common than epiblepharon and is more likely to require surgical correction. Corneal staining confirms lash abrasion and indicates the need for treatment.
Coloboma
A coloboma is a notch or cleft in the eyelid present from birth, usually affecting the upper lid at the junction of the medial and central thirds. Small colobomas may not cause corneal exposure; large defects require early reconstruction to protect the cornea from exposure keratopathy. Eyelid colobomas may be isolated or associated with Goldenhar syndrome (oculo-auriculo-vertebral spectrum), which includes ear and vertebral anomalies.
Distichiasis
Distichiasis is an accessory row of eyelashes arising from the meibomian gland orifices, directed toward the cornea. The lashes are typically fine and soft; many patients are asymptomatic. Symptomatic cases with corneal irritation or staining are treated with cryotherapy to the posterior lamella (freeze-thaw cycles to ablate aberrant follicles), electrolysis, or lid-splitting procedures for localized areas.
Schedule a Consultation
Contact us to discuss your concerns and learn about treatment options.